 CORBIS, ROBIN BARTHOLICK
CORBIS, ROBIN BARTHOLICK
Sometimes the human body turns on itself. Cancer and autoimmune diseases involve some form of physiological revolt, when a body’s own cells and molecules rise up to bring about its undoing. Patients suffering from most of these disorders have a variety of treatment options, formulated from a mechanistic understanding of the molecular roots of the diseases. But in acute pancreatitis, a largely untreatable disease, the exocrine cells of the pancreas—whose role is to manufacture enzymes that are normally mixed with bile from the liver to digest dietary proteins and fats—malfunction and digest themselves and surrounding tissues and organs in a more mysterious fashion. Acute pancreatitis is usually preceded either by heavy alcohol consumption or by blockage of the duct that unites the common bile duct and the pancreatic duct with hardened lumps of bile called gallstones. Searing pain, vomiting, fever, internal bleeding, multiple organ failure, and...
Acute pancreatitis involves an act of molecular cannibalism. A protease called trypsin, produced by the pancreas’s acinar cells, is normally released into the pancreatic duct as a proenzyme, before travelling to the gut, where it’s activated to break down the protein molecules in the food we eat. Inactive pancreatic proteases are packaged into small organelles called zymogen granules (ZGs), which are clustered inside the acinar cells. They are normally in a chemically inactivatable form, cemented together by calcium. But when acute pancreatitis strikes, the packaged enzymes become activated, break down the granules, and are then free in the cytoplasm, where they begin to digest the pancreas from the inside out.
For all that is known about the cellular progression of acute pancreatitis, its molecular triggers have remained cloaked in mystery. And neither science nor medicine has yet offered answers for how to treat the sometimes fatal disease. For more than 200,000 acute pancreatitis patients admitted to hospitals in the United States each year, that means relying only upon pain-relief drugs, intravenous fluid replacement, antibiotics, and changes in diet to stave off the serious internal damage that the disease can wreak.[1. M.W. Russo et al., “Digestive and liver disease statistics, 2004,” Gastroenterology, 126:1448-53, 2004.]
But recently, new light has been shed on the pathobiology of acute pancreatitis, from which prevention and treatment opportunities are emerging.
It starts with calcium
One hypothesis to explain the premature protease activation occurring in acute pancreatitis is that release of calcium ions from the ZGs allows their internal granular matrix to dissolve, making the enzymes activatable—the crucial step in initiating the disease. Trypsin activation depends on substantial release of intracellular calcium (Ca2+) from internal stores, followed by an influx of additional Ca2+ from the extracellular space.

It is also clear that as trypsin activation occurs, so does intracellular vacuolization—an event that involves the transformation of ZGs into empty-looking, misshapen vacuoles.[2. O.H. Petersen et al., “Fatty acids, alcohol and fatty acid ethyl esters: toxic Ca2+ signal generation and pancreatitis,” Cell Calcium, 45:634-42, 2009.] This never happens under normal conditions, when trypsin secretion occurs in response to food intake. Normal secretion occurs when ZGs fuse with the part of the cell membrane that lines the acinar lumen, forming a pore between the lumen and the interior of the ZGs through which the enzymes can pass and enter the pancreatic duct. This normal secretion process, like the abnormal trypsin activation that is a hallmark of pancreatitis, is also triggered by a rise in Ca2+ concentration inside the acinar cells, which is due to release of Ca2+ from internal stores.
For years, researchers struggled to understand why the same intracellular rise in Ca2+ concentration that is required for normal secretion can also trigger self-destruction of the cell. The answer to this question awaited the development of new technology—high-resolution dynamic imaging of live cells—allowing the precise definition of different kinds of intracellular Ca2+ signals. In the early 1990s we pioneered the imaging, at high temporal and spatial resolution, of intracellular Ca2+ signals evoked by different strengths of pancreatic stimulants. We found that the kind of stimuli generated normally in response to food intake—low blood concentrations of the pancreas-stimulating hormone cholecystokinin (CCK) or of the neurotransmitter acetylcholine (released from nerve endings in the neighborhood of the acinar cells)—created short-lasting, repetitive spikes in the intracellular Ca2+ concentration that only occurred locally in the region surrounding the ZGs. In contrast, high and abnormal hormone or neurotransmitter concentrations evoked sustained global elevations of the intracellular Ca2+ concentration. This led to our hypothesis that short-lasting local Ca2+ elevations were “good” signals, causing normal secretion, whereas sustained global Ca2+ elevations were “bad” signals leading to intracellular protease activation and self-destruction.[3. O.H. Petersen, A.V. Tepikin, “Polarized calcium signalling in exocrine gland cells,” Annu Rev Physiol, 70:273-99, 2008.]
Of mice and men
All these mechanistic studies on Ca2+ and trypsin activation used pancreatic acinar cells isolated from mice.3 There was, for some time, uncertainty whether the mouse cells were good models for their human pancreatic counterpart. One study suggested that human pancreatic acinar cells, in contrast to those of mice, did not possess receptors for the hormone CCK. It was not easy to obtain good-quality, normal human pancreatic acinar cells as a by-product of surgery on pancreatic cancer patients, but after many years of work, in collaboration with surgeons at the Royal Liverpool Hospital, we managed to show that human pancreatic acinar cells actually work in essentially the exact same way as our mouse models, and do possess functional CCK receptors.[4. J.A. Murphy et al.,"Direct activation of cytosolic Ca2+ signaling and enzyme secretion by cholecystokinin in human pancreatic acinar cells," Gastroenterology, 135:632-41, 2008.]
It was clear to us that the next step would be to study, at high resolution, the kind of intracellular Ca2+ signals generated by the substances—bile acids and alcohol—that actually induce pancreatitis in patients.
Physiology meets molecular pathogenesis
Bile, containing acids that are important for the digestion process, is formed in the liver and stored in the gallbladder, which contracts and expels the substance into the bile duct when food enters the small intestine. Gallstones, jagged little crystals formed in the gallbladder and most commonly composed of cholesterol, are known to induce attacks of acute pancreatitis, either by obstructing the pancreatic duct through which digestive enzymes pass into the gut, or by blocking the duct that unites the common bile duct and the pancreatic duct. This obstruction can allow reflux of bile acids into the pancreas, causing pancreatic injury. Detailed mechanistic studies by our own group demonstrated that bile acids released Ca2+ inside the cells from two distinct stores—and, importantly, showed that one of these was in the ZGs (the other store was in the endoplasmic reticulum).[5. J.V. Gerasimenko et al., “Bile acids induce Ca2+ release from both the endoplasmic reticulum and acidic intracellular calcium stores through activation of inositol trisphosphate receptors and ryanodine receptors,” J Biol Chem, 281:40154-63, 2006.]
The risk of developing pancreatitis increases with increasing alcohol intake, but only a minority (<10 percent of those drinking excessive amounts of alcohol develop the disease.[6. S.J. Pandol et al., “Investigating the pathobiology of alcoholic pancreatitis,” Alcohol Clin Exp Res, 35:830-37, 2011.] We have shown in isolated pancreatic acinar cells that even very high concentrations of alcohol generally have only modest effects on cellular Ca2+ homeostasis. This was a very confusing finding, since it cast doubt on whether alcohol in itself was a significant risk factor. We were helped by early postmortem studies showing high concentrations of fatty acid ethyl esters (FAEEs) in the pancreases of patients intoxicated with alcohol at the time of death. These observations suggested that the combination of long-chain fatty acids and ethanol to generate FAEEs could be important for the induction of pancreatitis.
The crucial point was then to find out whether pathophysiologically relevant concentrations of FAEEs would cause Ca2+-dependent destruction of pancreatic acinar cells. There are always fatty acids around the acinar cells, but one would expect the synthesis of FAEEs, which occurs inside the acinar cells, to accelerate with higher blood levels of fatty acids, which are easily transported into those cells. In fact, it is well known that high lipid levels in the blood predispose to pancreatitis.2 It turned out that FAEEs did indeed cause massive intracellular Ca2+ release in normal pancreatic acinar cells, and that an important component of this release occurred from the ZGs,[7. J.V. Gerasimenko et al., “Pancreatic protease activation by alcohol metabolite depends on Ca2+ release via acid store IP3 receptors,” PNAS, 106:10758-63, 2009.] exactly as had happened in the case of stimulation with bile acids.5
In follow-up studies, using various pharmacological agents, we were able to selectively interfere with Ca2+ release from either the ZGs or the endoplasmic reticulum. It turned out that it was the release from the ZGs that was specifically correlated with intracellular protease activation.7 In collaboration with a Japanese group, led by Katsuhiko Mikoshiba at the RIKEN Brain Science Institute near Tokyo, we conducted gene-deletion studies that demonstrated that the most important pathway for the FAEE-induced Ca2+ release from the ZGs was a particular set of Ca2+ channels, known as inositol trisphosphate receptors (IP3R).7 Deleting the genes that code for those specific channels dramatically reduced FAEE-induced intracellular trypsin activation.7
The rate of generation of these dangerous FAEEs will obviously depend on the concentrations of alcohol and fatty acids, but also on the activity of a class of acinar cell enzymes called FAEE synthases, whose mechanism of control is yet unknown.
It’s puzzling that a small proportion of isolated pancreatic acinar cells do generate substantial sustained intracellular Ca2+ elevations in response to alcohol alone. It is not known whether this could be due to abnormally high intracellular levels of fatty acids, or whether certain acinar cells could, under some conditions, be particularly sensitive to the effects of alcohol in isolation. In any case, this finding led to further research on the effect of alcohol on isolated acinar cells in which the cell membrane had deliberately been partially destroyed (permeabilized cells), and these studies revealed an interesting protective mechanism.
Calmodulin, coffee, and therapy
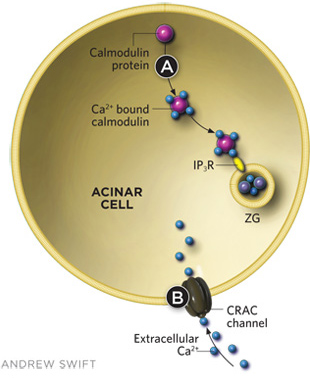
Some compounds have shown promise in reducing intracellular trypsin activation, which triggers acute pancreatitis. Calmodulin, a cytosolic calcium-binding protein found in normally functioning acinar cells, prevents ethanol from eliciting the release of substantial amounts of Ca2+ from intracellular stores, which can prematurely activate trypsin. Small peptide activators of calmodulin (such as CALP-3) can be added to the outside of acinar cells to help mobilize calmodulin and prevent the toxic effects of high alcohol concentrations. Calmodulin helps prevent trypsin activation by binding to inositol trisphosphate receptors (IP3Rs), which inhibits Ca2+ release from ZGs. Caffeine has been shown to have similar inhibitory effects on IP3Rs in ZG membranes (A).There is also the potential for small molecule therapies that interfere with CRAC function and thus alter the influx of extracellular Ca2+ (B).
Normally functioning pancreatic acinar cells have built-in protection from the effects of alcohol, a cytosolic calcium-binding protein called calmodulin that prevents ethanol from eliciting the release of substantial amounts of Ca2+ from intracellular stores.[8. J.V. Gerasimenko et al., “Calmodulin protects against alcohol-induced trypsinogen activation elicited via Ca2+ release through IP3 receptors,” PNAS, 108:5873-78, 2011.] When we washed calmodulin out of isolated, permeabilized acinar cells, we found that alcohol had a strong and acute toxic effect, while reintroduction of calmodulin at normal intracellular concentrations protected the cells from damage. We were particularly excited by the fact that a membrane-permeable small peptide activator of calmodulin (CALP-3), when added to the outside of permeabilized, isolated cells, prevented the toxic effects of even very high alcohol concentrations.8
Recent data indicating that trypsin activation is Ca2+-dependent suggest that reducing the activity of those Ca2+-release pathways may be help limit damage caused by pathological stimuli such as alcohol, alcohol–fatty acid metabolites, and bile acids. There are several subtypes of the crucial IP3R channels, and deletion of just one subtype does not affect normal stimulus-secretion coupling, but does markedly reduce intracellular trypsin activation induced by FAEEs.8
One compound recently shown also to achieve such inhibition is caffeine, which interferes with the opening of IP3R channels3,[9. O.H. Petersen, R. Sutton, “Ca2+ signalling and pancreatitis: effects of alcohol, bile and coffee,” Trends Pharmacol Sci, 27:113-20, 2006.] and reduces cytosolic Ca2+ signal generation in response to both FAEEs and bile acids. Caffeine has also been shown to reduce the probability of ethanol-induced pancreatitis in a clinical study.9 But caffeine’s potential as a therapeutic is unfortunately limited by its relatively low affinity for IP3Rs, so that the protective effect is only moderate.9 Nevertheless, it would appear that having a cup of strong coffee at the end of a rich meal that has been washed down with alcohol might be a good idea.
Other calcium channels—such as the CRAC channel, which allows the influx of extracellular Ca2+ in response to the release of Ca2+ from intracellular stores—may be important in acute pancreatitis, and could be potential therapeutic targets. As we expand our understanding of acute pancreatitis’s etiology, we come closer to providing preventive and therapeutic methods that precisely target molecular drivers of the disease, and ending the suffering it causes.
Ole H. Petersen is a Medical Research Council Professor and Director of the School of Biosciences at Cardiff University, Wales, UK. Both Oleg V. Gerasimenko and Julia V. Gerasimenko also work in the university’s School of Biosciences—he as a reader, she as a lecturer.
This article is adapted from a review in F1000 Medicine Reports, DOI:10.3410/M3-15.
Interested in reading more?




